
Optimal IGC
Quick Start
My personal encounters with the IGC literature suggested that much of it was of relatively low quality, but I did not know why. More careful study showed that there are many ways for IGC to go wrong if you don't attend to the minor details.
IGC seems so simple. Systematically inject known probes down a column of interest and from their retention times (and, in some cases, peak shapes) calculate the values of interest. In principle anyone can (and many people have done so) re-purpose a standard GC apparatus to get a "good enough" IGC setup.
But it is all too obvious from the literature that many "good enough" IGC setups are, in fact, not good enough. There are, unfortunately, lots of bad IGC data out there as I can personally attest to. Indeed, I have created (but not published!) plenty of bad data of my own which forced me to think hard about what an optimal IGC setup should be.
The right set of probes
For some simpler experiments, just having hexane, heptane, octane and nonane is good enough. But there are far too many experiments that are trying to generate broader understanding of surfaces by using, say, a set of alkanes then a set of alcohols - one providing "non-polar" and the other providing "polar" information. But if you are going to inject, say, 8 different probes, you get a far richer store of information by injecting a wide range of chemical functionalities. 4 alkanes and 4 alcohols is a wasted opportunity. The information from hexane, toluene, chloroform, MEK, acetonitrile, ethyl acetate, pyridine and ethanol will be far richer because there is a greater range of van der Waals, polar and H-bonding interactions.
Infinite dilution
The apps described here all make the assumption that the probe molecules interact only with the stationary phase and not with each other, i.e. so-called infinite dilution measurements. This requires a short, sharp, low-level injection of the probe molecule which in turn requires a fast, accurate injector system and a good, sensitive, detector.
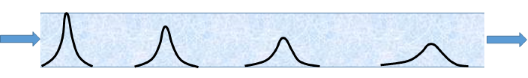 Even with an ideal injection there is a problem. The concentration at the start is relatively high because all the molecules are in a sharp peak. As the front migrates down the column, two effects change the probe concentration.
Even with an ideal injection there is a problem. The concentration at the start is relatively high because all the molecules are in a sharp peak. As the front migrates down the column, two effects change the probe concentration.
- The peak widens via diffusion driven by concentration gradients (broadening is fast at first as the concentration gradients are high) and by fluid dynamical effects, so the local concentrations are reduced
- The absorption on the support necessarily reduces the concentration in the gas phase,
Therefore, the conditions are closer to infinite dilution towards the end of the column than at the start! Analysis of the data must take this effect into account. For a given packing, the dilution of a non-absorbing probe will be less than that of an absorbant probe so what may be infinitely dilute for one probe may not be for another.
Finite pulse
An alternative methodology (not described in the apps here) injects a controlled square "pulse" of probe molecules and analyses the distortion away from the square shape at the detector. The optimal IGC equipment allows these contradictory capabilities: a short, sharp peak for infinite dilution and a controlled square pulse for finite concentration work. In both cases, the systems should be computer controlled to allow accurate timing analysis.
Perfect peak shape
The shape of the peak at the detector provides lots of extra information for analysis. So we need the shape to depend strongly on the surface we are analysing and be relatively free from artefacts from the IGC setup.
The shape of the injector pulse has already been discussed. This leaves three other areas for optimisation.
- Input/output tubing should have minimal effect. The physics of flow in tubing is well-known and the effects on peak shape can, in principle, be de-convoluted. But de-convolution is never perfect, so the tubing shape/length should be optimised for minimum perturbation. Remember, however, that some reasonable length is required to provide thermal isolation if there is a temperature difference between injection port and column.
- Dead space in the column is, of course, highly undesirable. Because different IGC problems involve different amounts of material in the column, this naturally means that column diameter and length should be chosen to fit the problem at hand. Never use a "one size fits all" column because you will either have too little packing to be meaningful or have empty space in the column which contributes strongly to peak broadening.
- Different samples pack in different ways and there will be different effects on back pressure and on peak shape. Here there is a mixture of science and art. Packing with tapping, vibration, ultrasonics are options with different trade-offs and the more important peak shape is to your analysis, the more you have to optimise the packing process.
Tuneable flow rates
The gas flow rate is important for any GC and is generally under good control. For IGC it is important for the flow rate to be under good computer control because it is often necessary to vary flow rates in a systematic manner, for example to determine if the probe molecules have had time to equilibrate with the system being analysed and, of course, measurement of diffusion coefficients depends on a systematic change of flow rate.
The right oven and detector
The ovens in modern GCs are generally excellent with good, accurate temperature control. Detectors are also likely to be good. The only issue with ovens is that a carelessly large oven might mean unnecessarily long input/output tube lengths (peak broadening) and/or long equilibration times for those experiments that measure the temperature dependence of retention times and peak shapes.
The range of IGC analyses would be extended if ovens were available at sub-ambient conditions, but these are less common. For example, pharma "excipients" with a higher vapour pressure will not remain for long on the support with the flow of gas. At sub-ambient conditions, the vapour pressure might be low enough to enable meaningful analyses.
Intelligent experiments
Getting meaningful data from IGC requires meaningful experiments which, in turn, require a thoughtful approach to what you are doing. For example, the measurement of HSP values is based on the assumption that the probe molecules have equilibrated with the material on the support and that the support itself is "neutral". To get proper equilibration may take time, so the flow rate of the experiment is important. Too low a rate and your throughput is unacceptably small. Too high a rate and the data are worthless. So at the very least, some tests with a couple of probes (high and low compatibility) at at least two flow rates will be necessary to see if the assumptions are valid.
Whichever analysis you are doing, it requires a similar thought process: think through the assumptions behind the algorithms used to obtain the data, then plan and test to make sure that your assumptions are valid whilst maintaining a reasonable experimental throughput.
Data analysis
The apps here are only indications of what can be done with IGC and focus mostly on retention time. The diffusion app simply uses peak width. But there is a lot to be learned from peak shapes. At a very minimum they provide insights into the general quality of your setup and for many analyses (such as with finite concentration pulses) peak shape is key.
So the optimal system requires data analysis software that can provide not only numbers from the basic analysis (via retention times and/or peak shapes) but some assessment of the quality of those numbers using other aspects of the data, including comparison with "neutral" test probes that provide information about the general setup - assuming, of course, that the test probe really is neutral.
Bad data, bad science, bad software
Because of my interest in HSP and because IGC measurements held so much promise, I have analysed many datasets using the algorithm described in the app, but using a more sophisticated version within the HSPiP program. For some datasets (such as those obtained with great care by Munk and by Voelkel), the fitting and the values have made a lot of sense. For others, the results were nonsense. The hard work of analysing the nonsense results meant (embarrasingly) that I found a bug in my own software. This happened to have no effect on datasets such as those of Munk but had a small, but significant, effect on other datasets. Fixing the bug was necessary, but the "nonsense" sets were still nonsense. Some of those sets were, on close scrutiny, done under poorly conceived experimental plans (e.g. with 5 alkanes, 4 alcohols and a couple of other probes), or done, for example, with no attention to flow rate so equilibrium was probably not attained. In all such cases the results are basically meaningless. But I had access to a a number of high quality datasets where the results were still nonsense. Having eliminated all other assumptions, the remaining big assumption was that the support material was "neutral". Repeating the experiments on a different support material suddenly gave sensible results. Clearly, the other support had been anything other than neutral.
Your own experiences will be different in detail. But the process is the same. Are the data bad for some reason? Is the science bad for some reason? Is the software faulty? In other words, IGC requires you to be a good scientist and question your assumptions.