
Solubility Effects
Quick Start
This is a very old page created when I had no way of appifying ideas in crystallization. It's a summary of what (little) I knew some years ago, but it still captures a few ideas in easy form. Refer to the real apps to know more!
 Knowing the solubility of a compound is not as easy as it sounds. Solubility expert
Paul Mahon from iFormulate has kindly provided this guide to many of the issues surrounding solubility. The original version can be found on the
iFormulate site.
Knowing the solubility of a compound is not as easy as it sounds. Solubility expert
Paul Mahon from iFormulate has kindly provided this guide to many of the issues surrounding solubility. The original version can be found on the
iFormulate site.
Most of the science needed to predict, control and define the stability of solutions was defined by scientists of the Victorian era such as Ostwald and Arrhenius. However there is widespread puzzlement and confusion about solubility. One reason is that solubility and stability studies are still sometimes regarded as trivial and so are not properly thought out and executed.
Measuring solubility
The solubility of a material is the concentration of the solute in a steady state, saturated solution at any given temperature. The solubility of a solid generally increases with increasing temperature and is often (but not always) greater when the melting point is lower and the latent heat of melting is smaller - as discussed in the Ideal Solubility app.
In practice real-world solubility measurements often measure only the apparent solubility, because the measurements are confused by other factors:
- Supersaturation: Simply dissolving a solute in solvent, especially if you use agitation, heat or ultrasound will usually create an thermodynamically unstable “supersaturated” solution.
- Purity: generally speaking the purer the (solute) material is then the lower its solubility will be.
- Measurement method: The apparent solubility can sometimes vary depending on how you measure it.
- Polymorphic form: Most solutes will have many different polymorphic (crystal) forms, with widely differing solubilities.
- Particle size: Very small particles can be (at least for a short time) much more soluble than larger particles.
- Nucleation & growth: In the absence of heterogeneous nucleation sites metastable solutions can be indistinguishable from true solutions.
- Chemical stability: Reactions can occur between the solutes, solvents or environment.
The last factor is outside the scope of this page. The others are discussed in turn.
Supersaturation
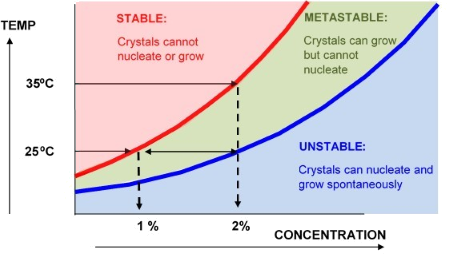 The supersolubility limit is the concentration at which spontaneous crystallisation occurs in the absence of heterogeneous nuclei. Heterogeneous nuclei can be anything from scratches on the container walls to bits of foreign matter, dust etc. The metastable region in the diagram is the area lying between the solubility and supersolubility concentrations. In this area, a crystal will grow but spontaneous nucleation will not occur (over reasonable time periods).
The supersolubility limit is the concentration at which spontaneous crystallisation occurs in the absence of heterogeneous nuclei. Heterogeneous nuclei can be anything from scratches on the container walls to bits of foreign matter, dust etc. The metastable region in the diagram is the area lying between the solubility and supersolubility concentrations. In this area, a crystal will grow but spontaneous nucleation will not occur (over reasonable time periods).
In the case above the solute has an equilibrium solubility of 1% at 25⁰C, and 2% at 35⁰C. A clean 2% solution cooled from 35⁰C to 25⁰C will still appear to be stable because there are no crystals to grow. However, the introduction of any nuclei would cause a rapid precipitation of half the material from solution. For this reason we do not normally use heat, ultrasound or high shear when measuring solubilities and we would usually ensure that there are nucleation sites present in the vessel.
Purity
High purity materials tend to have lower solubility than slightly impure materials, this is because impurities can get into the crystal lattice causing dislocations which change the chemical potential of the solid – in the same way that impurities lower the melting point. However, different types of impurity can have different effects, some can increase solubility but others can decrease solubility. For this reason if the source, grade or purity of the solute or solvent is changed in any way, the solubility and/or stability of the solute will need re-assessing.
Measurement Method
This is particularly important if the solute is a mixture – either by design or because it contains impurities. If we make a supersaturated mixture of solute and solvent and then measure how much is in solution we will be measuring the most soluble components of the mixture. However, if we take clean solvent and add solute to it until we see material out of solution then we would be measuring the least soluble component of the mixture.
Polymorphic Form
Polymorphism is all about minimising free energy. The free energy is a mix of a (positive) surface energy term from γ, surface energy and A, the area of the crystals, and a (negative) term mechemical potential term based on θ the change of chemical potential and N the number of moles.
ΔG=γA-θN
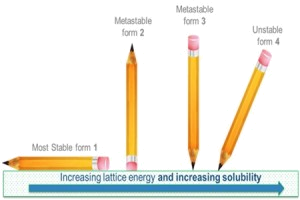 The issue here is that different crystalline forms (polymorphs) have different chemical potentials. The one with the largest change in potential (i.e. the most stable form) is the least soluble and is guaranteed to be the crystal form found after an infinite time. The problem is that we aren't guaranteed
any crystals (i.e. supersaturation), and the crystal that appears might be kinetically favoured even though it is not the lowest-energy polymorph. So in practice what this means is that the supplied solute may often be in a more soluble polymorphic form, but it may change to a less soluble form when in solution via a process known as solvent mediated polymorphic phase transformation.
The issue here is that different crystalline forms (polymorphs) have different chemical potentials. The one with the largest change in potential (i.e. the most stable form) is the least soluble and is guaranteed to be the crystal form found after an infinite time. The problem is that we aren't guaranteed
any crystals (i.e. supersaturation), and the crystal that appears might be kinetically favoured even though it is not the lowest-energy polymorph. So in practice what this means is that the supplied solute may often be in a more soluble polymorphic form, but it may change to a less soluble form when in solution via a process known as solvent mediated polymorphic phase transformation.
If we measure the wrong (less stable) polymorph we will always get too high a result for our solubility. For this reason we need to convert the solute to its most stable polymorphic form. This can usually be achieved by temperature cycling a saturated solution. The temperature cycling conditions required depend upon the solvent but usually a -10°C to +40°C daily cycle for a few days will suffice.
Particle Size
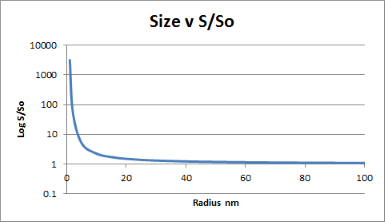 The same Gibbs free energy equation tells us that solubility is dependent on particle size, i.e. their surface area to volume ratio. It only really makes a difference when the particles are very small <50nm, as shown. The equation involved is Ostwald-Freundlich. Where γ is the surface energy, r is the radius and Vm is the molar volume then the ratio of solubility at radius r and the solubility of large crystals So is given by:
The same Gibbs free energy equation tells us that solubility is dependent on particle size, i.e. their surface area to volume ratio. It only really makes a difference when the particles are very small <50nm, as shown. The equation involved is Ostwald-Freundlich. Where γ is the surface energy, r is the radius and Vm is the molar volume then the ratio of solubility at radius r and the solubility of large crystals So is given by:
ln(S/S0)=2(γ/r)(Vm/RT)
Again the problem is that measuring solubility of very small particles will give too high a result, and the solution will be unstable long-term because a process called Ostwald ripening will occur - the process (driven by surface energy) where smaller crystals get smaller and larger crystals get larger. The small crystals may well dissolve easily, but at some later stage large crystals will grow because they will have lower solubility and the solution will be supersaturated with respect to the larger crystals. For this reason we need to convert sub-micron solutes to larger crystal sizes via Ostwald ripening. This can also usually be achieved by temperature cycling a saturated solution as above.
Nucleation & Growth
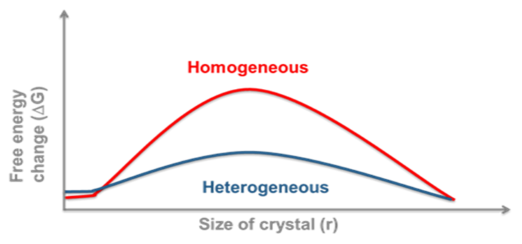 Nucleation and growth are thermodynamically distinct processes, with nucleation being the initial formation of a crystallite of a critical size and then growth being the subsequent exponential increase in the crystal size. Again, this has practical implications. Homogeneous nucleation (in the absence of any nucleation sites) usually has a much higher free energy barrier than heterogeneous nucleation. The implication is that when stability or solubility is measured in clean laboratory glassware for instance, then a supersaturated solution might be likely. This may act like a true solution until the introduction of foreign nuclei results in sudden catastrophic crystallisation. On an industrial scale there will almost always be foreign bodies or rough surfaces to act as a scaffold for nucleation. Famously, if a production site accidentally produces the wrong polymorph, these seed crystals might be impossible to remove and the site will forever produce the wrong crystal type. In the testing lab it is vital to ensure that sufficient nucleation sites are available, this can be from scratching glassware or metal containers or by the intentional introduction of seed crystals.
Nucleation and growth are thermodynamically distinct processes, with nucleation being the initial formation of a crystallite of a critical size and then growth being the subsequent exponential increase in the crystal size. Again, this has practical implications. Homogeneous nucleation (in the absence of any nucleation sites) usually has a much higher free energy barrier than heterogeneous nucleation. The implication is that when stability or solubility is measured in clean laboratory glassware for instance, then a supersaturated solution might be likely. This may act like a true solution until the introduction of foreign nuclei results in sudden catastrophic crystallisation. On an industrial scale there will almost always be foreign bodies or rough surfaces to act as a scaffold for nucleation. Famously, if a production site accidentally produces the wrong polymorph, these seed crystals might be impossible to remove and the site will forever produce the wrong crystal type. In the testing lab it is vital to ensure that sufficient nucleation sites are available, this can be from scratching glassware or metal containers or by the intentional introduction of seed crystals.
